【紹介する論文はCC-BYになっており、画像や文章の商用引用も可能な論文です。】
https://s100.copyright.com/AppDispatchServlet?publisherName=ELS&contentID=S0092867425010372&orderBeanReset=true&orderSource=Phoenix
本日紹介する論文は先日Cellに発表されたRAEFISHという新しい空間トランスクリプトーム技術に関する論文です。網羅性やコストといったこれまでの技術の問題点を様々な工夫で克服しており、今後この技術が空間トランスクリプトームの定番になるかもしれません。
管理人も理解するのに時間がかかってしまい記事作成が遅くなってしまいましたが、現状の空間トランスクリプトーム技術についても触れつつ解説したいと思います。
論文のタイトルは「Sequencing-free whole-genome spatial transcriptomics at single-molecule resolution」です(https://www.cell.com/cell/fulltext/S0092-8674(25)01037-2)。
これまでの空間トランスクリプトーム技術について
空間トランスクリプトーム技術は細胞や組織において、空間情報を保持しつつ遺伝子発現を評価する手法のことです。Nature誌から2020年のMethod of the yearにも選ばれており、ヒト組織を用いた病態解明や細胞内のRNAの制御の解明などに用いられるようになっています。
現在用いられている空間トランスクリプトーム技術を分類すると大きく分けて、
・空間をBarcodeで識別されるスポットやビーズに分割し、その中でRNAをBarcode付きオリゴでCaputureすることで、シークエンスした際にRNAがどの場所由来かわかるもの。VisiumやSlide-seq, Stereo-seqなど。網羅性が高いが、空間分解能はビーズやスポットの大きさに依存する。
・画像ベースに細胞内の一分子のRNAをFISH法やin situシーケンス (ISS)により検出するもの。seq-FISH, MERFISH, Xenium, STAR-map (ISS)など。一分子レベルと空間分解能が高いが、網羅性が低い。
というように空間分解能と網羅性がある種のトレードオフになっており、研究者は目的に合わせて必要な技術を使用していました。
今回はこの一分子レベルの空間解析を全ゲノムスケールで、しかも低コストで行えるようになったという論文です。まさにいいとこどりですね。
そんなうまい話があるのかと思ったのですが(笑)、理屈が分かるとなるほど~となりました。
これまでの技術の融合
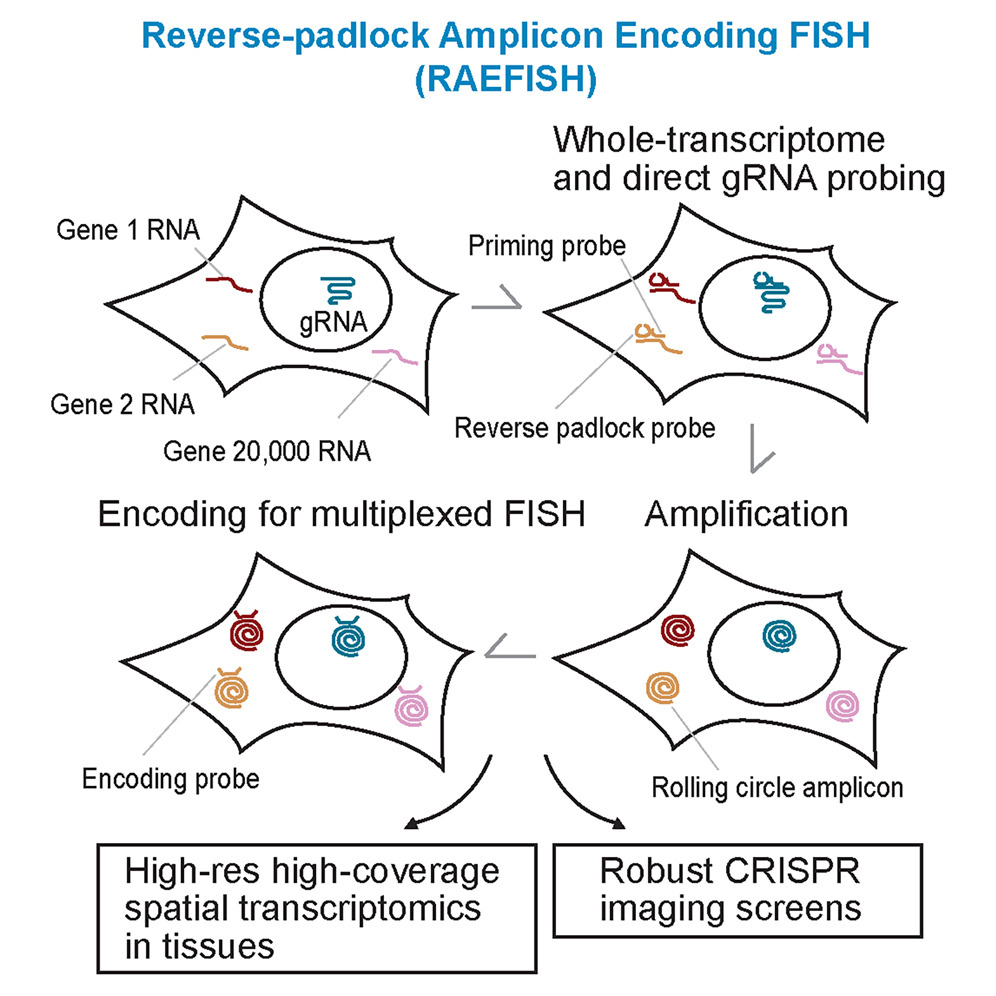
このRAEFISHはSTARmapの技術によりRNAをin situで増幅させて、MERFISHの技術で識別するというコンセプトです。さらにそのProbeの設計にもうひと工夫してあり、STAR-mapと違って低コストでできるようにしています。
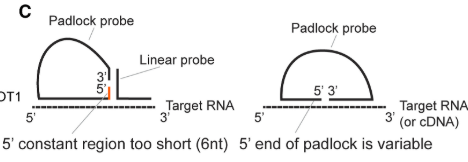
まずSTARmapの部分ですが、目的のRNAに特異的に結合するPriming probeとPadlock probeをハイブリダイゼーションします。Padlockとは南京錠という意味で、Padlock probeは両端を閉じることで環状DNAになります。あとで触れますがこのPadlock probeをSplint oligoで閉じることで、低コスト化を実現しており、この改良されたものをReverse padlock probeと命名しています。
こうしてできた環状のDNAを鋳型に、一本鎖DNAを連続的に合成することで同じ配列を何百回も増幅する、rolling circle amplification (RCA)を行うことで、Probeが結合した特異的配列が局所で増幅します(Fig.1A 上段)。
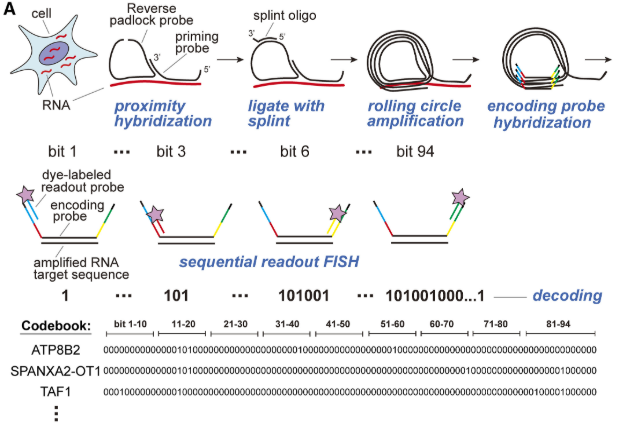
ここからがMERFISHの応用になるのですが、この増幅された配列に対して4種類のBarcodeがついたencoding probeをハイブリダイゼーションします。このBarcodeは全体で94種類あり、その組み合わせによりそれぞれのRNAを識別することが可能です。
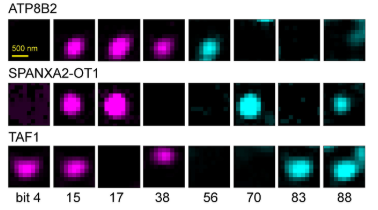
ここからこの94種類のBarcodeに対応する、2種類の蛍光でラベルされたProbeを順番にハイブリダイゼーションすることで、どのラウンドで光ったかの情報を基にRNAの種類を同定しています(1A中段)。
光った時を1として光らなかった時を0としており、例えばATP8B2は15番目と17番目と、38番目と56番目にの光ったものをこの遺伝子として同定していることになります(1A下段, 1D)。
理論上は94種類から4つの組み合わせで、94C4になるので300万通り近くを識別できることになります。ただ、実際は同時に光る遺伝子が多くなると正確性が低下するので、一ラウンドに光る遺伝子が全体の4%程度になるように組み合わせは制限されます。それでも全ゲノム2万遺伝子には十分な組み合わせになりますね。
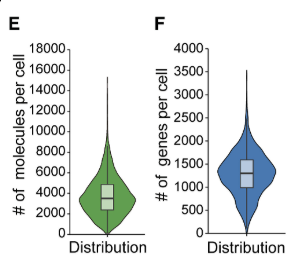
実際にCell lineで検出感度をみると、1細胞あたり平均して3749分子のRNAを検出し、1287種類の遺伝子を検出していました。これはSingle cell RNA-seqやStereo-seq, STARmapなどと同程度で、シーケンス型と同程度の検出感度を確保していました。。
MERFISHは一つのRNAに複数のProbeをあてることでシグナルを増幅しているのですが、そのためにProbeの種類が多くなりすぎて全ゲノムは難しいのと、Probeが複数あてられない短いRNAは検出できないのが欠点でした。そこをSTAR-mapの技術で標的配列を増幅することで、1種類のProbeで十分なシグナルを確保できるようにしたんですね。
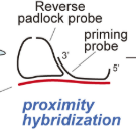
Reverse padlock probeでなぜ安くなるのか
この手法はコストが下がるための工夫もされています。
STAR-map(左)やXenium(右)で使用されているPadlock probeはその両端かもしくは一方が、標的のRNAに特異的な配列になっていました。このために自分たちでPCRしてProbeを増やすことができず、毎回合成するか購入しないといけないので金銭的な負担が大きくなっていました。
今回使われているReverse padlockでは、両端は共通の配列になっているのでPCRでまとめて増幅することが出来ます。そのためLibraryの費用を抑えることができ、5100ドルで2000回分の実験ができるそうです。Probe以外のもろもろの試薬のコストを入れても158ドルで一回の実験ができるらしく、STARmapの123倍安い!と本文中に書いてます(笑)。
実際に使ってみた
この技術を実際に使ってCell lineの細胞周期の同定や、マウスの肝臓や胎盤、リンパ節での遺伝子発現を評価して、これまで知られている組織の構造や細胞種を再現できており、また全ゲノムスケールで遺伝子発現を評価できていることを確認しています。
CRISPR screenとの併用
ここから面白かったのがCRISPR screenと併用することで、シーケンスフリーに各遺伝子をKOした際の表現型やRNAの局在の変化を評価できることを示していることです。CRISPR screenはgRNAをレンチウイルスで遺伝子導入して、一つの細胞で一つの遺伝子がKOされる細胞集団を用いて、網羅的に遺伝子機能を評価する手法です。
図のようにgRNAを認識するProbeを作成することで、内在性の遺伝子と同様にgRNAも検出しています。想定通り1つの細胞につき、1種類のgRNAが発現していることがほとんどであることも確認しています(Fig.1C)。
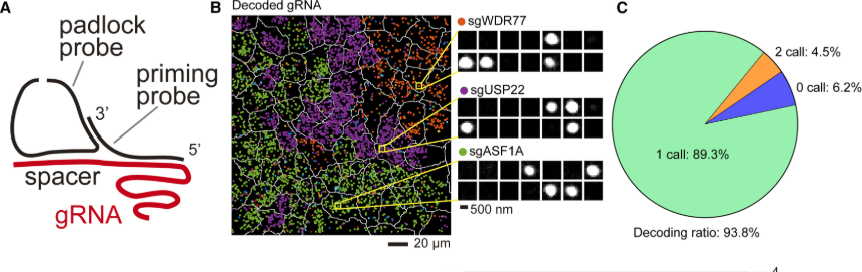
最後に500遺伝子程のノックアウトライブラリーを使って、ノックアウトされる遺伝子ごとのRNAの発現量の変化をシーケンスフリーでとらえることが出来ることを示しています。
感想とLimitation
空間トランスクリプトーム技術の一つの到達点が来たなと感じました。
理論通りであれば網羅性、空間分解能、コストの面で理想的な技術なのだろうと思います。
特に網羅性についてはこれまでFISH-seq系の技術は遺伝子セットを用意するためにHypothesis drivenになりがちだったので、Unbiaseに調べることが出来るのは大きな進歩と思います。ビッグデータになりそうなのでAIとも親和性が高そうですね。
Limitationとして、MERFISHと比較すると個別の遺伝子の検出感度は劣るようです。これは網羅性とトレードオフでしょうがないのかなと思います。
また、高発現の遺伝子だと信号が重なってしまうので、House keeping geneなどの遺伝子は外す必要があるらしく、こちらの方は問題かなと思いました。今後はExpansion microscopyで信号の混雑を緩和することを検討しているようです。個人的にはサイトカインのように刺激で急激に増える遺伝子をどの程度正確に評価できるのか気になりました。

コメント